


Next: BPTI Example
Up: Creating PSF Structure Files
Previous: Ordinary Usage
Contents
- topology
 file name
file name
Purpose: Read in molecular topology definitions from file.
Arguments: file name: CHARMM format topology file.
Context: Beginning of script, before segment. May call multiple times.
- alias residue alternate name real name
Purpose: Provide translations from residues found in PDB files to proper
residue names read in from topology definition files. Proper names
from topology files will be used in generated PSF and PDB files.
Arguments: alternate name: Residue name found in PDB file.
real name: Residue name found in topology file.
Context: Before reading sequence with pdb. May call multiple times.
- segment segment ID { commands }
Purpose: Build a segment of the molecule. A segment is typically a single
chain of protein or DNA, with default patches applied to the termini.
Segments may also contain pure solvent or lipid.
Arguments: segment ID: Unique name for segment, 1-4 characters.
commands: Sequence of commands in Tcl syntax to build the primary
structure of the segment, including auto, first, last, residue, pdb, etc.
Context: After topology definitions and residue aliases. May call multiple times.
Structure information is generated at the end of every segment command.
- auto
 angles
angles![$]$](img9.gif) dihedrals none
dihedrals none
Purpose: Override default settings from topology file for automatic generation of
angles and dihedrals for the current segment.
Arguments: angles: Enable generation of angles from bonds.
dihedrals: Enable generation of dihedrals from angles.
none: Disable generation of angles and dihedrals.
Context: Anywhere within segment, does not affect later segments.
- first patch name
Purpose: Override default patch applied to first residue in segment.
Default is read from topology file and may be residue-specific.
Arguments: patch name: Single-target patch residue name or none.
Context: Anywhere within segment, does not affect later segments.
- last patch name
Purpose: Override default patch applied to last residue in segment.
Default is read from topology file and may be residue-specific.
Arguments: patch name: Single-target patch residue name or none.
Context: Anywhere within segment, does not affect later segments.
- residue resid resname
Purpose: Add a single residue to the end of the current segment.
Arguments: resid: Unique name for residue, 1-5 characters, usually numeric.
resname: Residue type name from topology file.
Context: Anywhere within segment.
- pdb file name
Purpose: Extract sequence information from PDB file when building segment.
Residue IDs will be preserved, residue names must match entries in
the topology file or should be aliased before pdb is called.
Arguments: file name: PDB file containing known or aliased residues.
Context: Anywhere within segment.
- mutate resid resname
Purpose: Change the type of a single residue in the current segment.
Arguments: resid: Unique name for residue, 1-5 characters, usually numeric.
resname: New residue type name from topology file.
Context: Within segment, after target residue has been created.
- patch patch residue name segid:resid ...
Purpose: Apply a patch to one or more residues. Patches make small modifications to
the structure of residues such as converting one to a terminus, changing the
protonation state, or creating disulphide bonds between a pair of residues.
Arguments: patch residue name: Name of patch residue from topology definition file.
segid:resid: List of segment and residue pairs to which patch should be applied.
Context: After one or more segments have been built.
- multiply factor segid:resid:atomname ...
Purpose: Create multiple images of a set of atoms for use in locally enhanced sampling. The beta column of the output pdb file is set to 1...factor for each image. Multiple copies of bonds, angles, etc. are created. Atom, residue or segment names are not altered; images are distinguished only by beta value. This is not a normal molecular structure and may confuse other tools.
Arguments: factor:
segid:resid:atomname: segment, residue, or atom to be multiplied. If :resid is omitted the entire segment is multiplied; if :atomname is omitted the entire residue is multiplied. May be repeated as many times as necessary to include all atoms.
Context: After one or more segments have been built, all patches applied, and coordinates guessed. The effects of this command may confuse other commands.
- delatom segid resid atom name
Purpose: Delete one or more atoms. If only segid is specified, all atoms from
that segment will be removed from the structure. If both segid and
resid are specified, all atoms from just that residue will be removed.
If segid, resid, and atom name are all specified, just a
single atom will be removed.
Arguments: segid: Name of segment.
resid: Name of residue (optional).
atom name: Name of atom (optional).
Context: After all segments have been built and patched.
- resetpsf
Purpose: Delete all segments in from the structure. The topology definitions and
aliases are left intact.
Arguments:
Context: After one or more segments have been built.
- writepsf charmm x-plor file name
Purpose: Write out structure information as PSF file.
Arguments: charmm: Use CHARMM format (numbers for atom types).
x-plor: Use X-PLOR format (names for atom types), the default format required by NAMD.
file name: PSF file to be generated.
Context: After all segments have been built and patched.
- readpsf file name
Purpose: Read in structure information from PSF file and adds it to the structure.
It is an error if any segments in the PSF file already exist.
Arguments: file name: PSF file in X-PLOR format (names for atom types).
Context: Anywhere but within segment.
- alias atom residue name alternate name real name
Purpose: Provide translations from atom names found in PDB files to proper
atom names read in from topology definition files. Proper names
from topology files will be used in generated PSF and PDB files.
Arguments: residue name: Proper or aliased residue name.
alternate name: Atom name found in PDB file.
real name: Atom name found in topology file.
Context: Before reading coordinates with coordpdb. May call multiple times.
- coord segid resid atomname { x y z }
Purpose: Set coordinates for a single atom.
Arguments: segid: Segment ID of target atom.
resid: Residue ID of target atom.
atomname: Name of target atom.
{ x y z }: Coordinates to be assigned.
Context: After structure has been generated.
- coordpdb file name segid
Purpose: Read coordinates from PDB file, matching segment, residue and atom names.
Arguments: file name: PDB file containing known or aliased residues and atoms.
segid: If specified override segment IDs in PDB file.
Context: After segment has been generated and atom aliases defined.
- guesscoord
Purpose: Guesses coordinates of atoms for which they were not explicitly set.
Calculation is based on internal coordinate hints contained in toplogy
definition files. When these are insufficient, wild guesses are attempted
based on bond lengths of 1 Å and angles of 109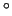 .
.
Arguments: None.
Context: After stucture has been generated and known coordinates read in.
- writepdb file name
Purpose: Writes PDB file containing coordinates. Atoms order is identical to
PSF file generated by writepsf (unless structure has been changed).
The O field is set to 1 for atoms with known coordinates, 0 for atoms
with guessed coordinates, and -1 for atoms with no coordinate data
available (coordinates are set to 0 for these atoms).
Arguments: file name: PDB file to be written.
Context: After structure and coordinates are complete.
Next: BPTI Example
Up: Creating PSF Structure Files
Previous: Ordinary Usage
Contents
namd@ks.uiuc.edu