




Next: Requirements
Up: Cartesian Coordinate Space
Previous: Dynamics Restarts
The dynamics Verlet statement sets up various parameters for molecular dynamics
as well as Langevin dynamics and then executes the specified
number of steps.
- DYNAmics VERLet
- { <dynamics-Verlet-statement> } END
is invoked from the main level of X-PLOR.
- <dynamics-Verlet-statement> :==
-
- ASCIi=<logical>
- writes a formatted (ASCII) coordinate
trajectory
file if ASCIi is TRUE;
if ASCIi is FALSE, writes an unformatted (binary) file.
In the former case,
a scale factor s and an offset o are applied to the
coordinates r (i.e.,
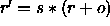 ); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor s and the offset o
results in positive
coordinates
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor s and the offset o
results in positive
coordinates
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
- CSEL=<selection>
- selects atoms that are
written to the coordinate trajectory file (default: all atoms except those
fixed by the constraints fix statement).
- FINAltemp=<real>
- determines the target temperature for the
velocity rescaling option (see ISCVel) (default: 298K).
- FIRSttemp=<real>
- assigns the temperature that determines the
initial velocity (default: 0K).
- FORMat=<string>
- provides FORTRAN format
for the ASCIi= TRUE option
(default: 12Z.6, hexadecimal).
- IASVelocity=<MAXWell|UNIForm|CURRent>
- determines the
mode of
initial velocity assignments
(default: zero velocities).
- IEQFrq=<integer>
- determines the frequency with which
the velocities are rescaled (default: 0). It works in conjunction
with FINAltemperature.
- ILBFrq=<integer>
- updates Langevin boundary between normal and
Langevin particles (default: 0). It works
in conjunction with RBUF and ORIGin.
- IPRFrq=<integer>
- prints energy statistics every IPRFrq steps
if not equal to zero (default: 50).
- ISCVel=<integer>
- determines the velocity rescale mode
(default: 0).
- ISVFrq=<integer>
- determines how often a restart file
is written to disk (default: 0).
- NPRInt=<integer>
- determines the frequency with which
the energy is printed to standard output (default: 1, i.e., at
every dynamics step).
- NSAVC=<integer>
- determines the frequency with which
the coordinates are written to the coordinate trajectory file (default: 0).
- NSAVV=<integer>
- determines the frequency with which
the velocities are written to the velocity trajectory file (default: 0).
- NSTEp=<integer>
- determines the number of steps. In the
case of a restart, the steps executed before writing the
restart file are included in NSTEP (e.g., if NSTEP=3000, but
1000 steps were run before writing the restart file, the program
will execute only 2000 steps) (default: 100).
- NTRFrq=<integer>
- determines the frequency with which
the center-of-mass motion is removed (default: 0).
- OFFSet=<real>
- provides offset o for the ASCIi=TRUE
option (default: 800).
- ORIGin=<vector>
- is the origin of the sphere of the Langevin
boundary (default: 0,0,0).
- RBUF=<real>
- specifies
the radius of the spherical boundary in a dynamics Verlet statement. Inside the
boundary, the particles are ``normal" molecular
dynamics particles; outside they are propagated as
Langevin particles (default: 0).
- RESTart=filename
- reads restart information from the named
file and restarts the dynamics (default: no restart).
- SAVE=<filename>
- specifies the name of the file
to which the program periodically writes the restart information (default:
none).
- SCALe=<real>
- provides a scale factor s for the
ASCIi=TRUE option (default: 10000).
- TBATh=<real>
- specifies
the temperature of the heatbath or the
target temperature of Berendsen's temperature-coupling method.
- TCOUpling=<logical>
- is a logical flag that turns on
Berendsen's temperature-coupling method. This flag works in
conjunction with TBATh (default: FALSE).
- TIMEstep=<real>
- assigns the time step
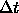 value for the finite-difference integration in psec
(default: 0.001 psec).
value for the finite-difference integration in psec
(default: 0.001 psec).
- TRAJectory=<file>
- provides the filename of the
coordinate trajectory file (default: none).
- VASCii=<logical>
- writes
a formatted (ASCII) velocity
trajectory file if VASCii is TRUE; if VASCii
is FALSE, an unformatted (binary) velocity
trajectory file is written.
In the former case,
a scale factor s and an offset o are applied to the
velocities r (i.e.,
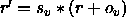 ); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor
); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor 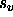 and the offset
and the offset 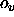 results in positive
velocities
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
results in positive
velocities
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
- VELOcity=<filename>
- names the velocity
trajectory file (default: none).
- VFORmat=<string>
- is the FORTRAN format
of the velocity trajectory file for the VASCii= TRUE option
(default: 12Z.6, hexadecimal).
- VOFFset=<real>
- provides an offset
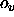 for the VASCii= TRUE
option (default: 300).
for the VASCii= TRUE
option (default: 300).
- VSCAle=<real>
- provides a scale factor
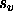 for the
VASCii=TRUE option (default: 10000).
for the
VASCii=TRUE option (default: 10000).
- VSEL=<selection>
- selects atoms that
are written to the velocity trajectory file (default: all atoms except
those fixed by the constraints fix statement).
Next: Requirements
Up: Cartesian Coordinate Space
Previous: Dynamics Restarts
Web Manager
Sat Mar 11 09:37:37 PST 1995