#include <molecule.h>
Inheritance diagram for Molecule
Public Methods | |
| Molecule () | |
| Molecule (const Molecule &) | |
| Molecule (StateIn &) | |
| Molecule (const Ref< KeyVal > &input) | |
| The KeyVal constructor. | |
| virtual | ~Molecule () |
| Molecule& | operator= (const Molecule &) |
| void | add_atom (int Z, double x, double y, double z, const char *=0, double mass=0.0, int have_charge=0, double charge=0.0) |
| Add an AtomicCenter to the Molecule. | |
| virtual void | print (std::ostream &=ExEnv::out()) const |
| Print information about the molecule. | |
| virtual void | print_parsedkeyval (std::ostream &=ExEnv::out(), int print_pg=1, int print_unit=1, int number_atoms=1) const |
| int | natom () const |
| Returns the number of atoms in the molcule. | |
| int | Z (int atom) const |
| double& | r (int atom, int xyz) |
| const double& | r (int atom, int xyz) const |
| double* | r (int atom) |
| const double* | r (int atom) const |
| double | mass (int atom) const |
| const char* | label (int atom) const |
| int | atom_at_position (double *, double tol=0.05) const |
| Takes an (x, y, z) postion and finds an atom within the given tolerance distance. More... | |
| int | atom_label_to_index (const char *label) const |
| Returns the index of the atom with the given label. More... | |
| double* | charges () const |
| Returns a double* containing the nuclear charges of the atoms. More... | |
| double | charge (int iatom) const |
| Return the charge of the atom. | |
| double | nuclear_charge () const |
| Returns the total nuclear charge. | |
| void | set_point_group (const Ref< PointGroup > &, double tol=1.0e-7) |
| Sets the PointGroup of the molecule. | |
| Ref<PointGroup> | point_group () const |
| Returns the PointGroup of the molecule. | |
| Ref<PointGroup> | highest_point_group (double tol=1.0e-8) const |
| Find this molecules true point group (limited to abelian groups). More... | |
| int | is_axis (SCVector3 &origin, SCVector3 &udirection, int order, double tol=1.0e-8) const |
| Return 1 if this given axis is a symmetry element for the molecule. More... | |
| int | is_plane (SCVector3 &origin, SCVector3 &uperp, double tol=1.0e-8) const |
| Return 1 if the given plane is a symmetry element for the molecule. More... | |
| int | has_inversion (SCVector3 &origin, double tol=1.0e-8) const |
| Return 1 if the molecule has an inversion center. | |
| int | is_linear (double tolerance=1.0e-5) const |
| Returns 1 if the molecule is linear, 0 otherwise. | |
| int | is_planar (double tolerance=1.0e-5) const |
| Returns 1 if the molecule is planar, 0 otherwise. | |
| void | is_linear_planar (int &linear, int &planar, double tol=1.0e-5) const |
| Sets linear to 1 if the molecular is linear, 0 otherwise. More... | |
| SCVector3 | center_of_mass () const |
| Returns a SCVector3 containing the cartesian coordinates of the center of mass for the molecule. | |
| double | nuclear_repulsion_energy () |
| Returns the nuclear repulsion energy for the molecule. | |
| void | nuclear_repulsion_1der (int center, double xyz[3]) |
| Compute the nuclear repulsion energy first derivative with respect to the given center. | |
| void | nuclear_efield (const double *position, double *efield) |
| Compute the electric field due to the nuclei at the given point. | |
| void | nuclear_charge_efield (const double *charges, const double *position, double *efield) |
| Compute the electric field due to the given charges at the positions of the nuclei at the given point. | |
| void | symmetrize (double tol=0.5) |
| If the molecule contains only symmetry unique atoms, this function will generate the other, redundant atoms. More... | |
| void | symmetrize (const Ref< PointGroup > &pg, double tol=0.5) |
| Set the point group and then symmetrize. | |
| void | cleanup_molecule (double tol=0.1) |
| This will try to carefully correct symmetry errors in molecules. More... | |
| void | translate (const double *r) |
| void | move_to_com () |
| void | transform_to_principal_axes (int trans_frame=1) |
| void | transform_to_symmetry_frame () |
| void | print_pdb (std::ostream &=ExEnv::out(), char *title=0) const |
| void | read_pdb (const char *filename) |
| void | principal_moments_of_inertia (double *evals, double **evecs=0) const |
| Compute the principal moments of inertia and, possibly, the principal axes. | |
| int | nunique () const |
| Return information about symmetry unique and equivalent atoms. | |
| int | unique (int iuniq) const |
| Returns the overall number of the iuniq'th unique atom. | |
| int | nequivalent (int iuniq) const |
| Returns the number of atoms equivalent to iuniq. | |
| int | equivalent (int iuniq, int j) const |
| Returns the j'th atom equivalent to iuniq. | |
| int | atom_to_unique (int iatom) const |
| Converts an atom number to the number of its generating unique atom. More... | |
| int | atom_to_unique_offset (int iatom) const |
| Converts an atom number to the offset of this atom in the list of generated atoms. More... | |
| int | n_core_electrons () |
| Return the number of core electrons. | |
| int | max_z () |
| Return the maximum atomic number. | |
| Ref<AtomInfo> | atominfo () const |
| Return the molecules AtomInfo object. | |
| void | save_data_state (StateOut &) |
| Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. More... | |
Protected Methods | |
| void | init_symmetry_info (double tol=0.5) |
| void | clear_symmetry_info () |
| void | clear () |
Protected Attributes | |
| int | natoms_ |
| Ref<AtomInfo> | atominfo_ |
| Ref<PointGroup> | pg_ |
| Ref<Units> | geometry_units_ |
| double** | r_ |
| int* | Z_ |
| double* | charges_ |
| int | nuniq_ |
| int* | nequiv_ |
| int** | equiv_ |
| int* | atom_to_uniq_ |
| double* | mass_ |
| char** | labels_ |
It has a KeyVal constructor that can create a new molecule from either a PDB file or from a list of Cartesian coordinates.
The following ParsedKeyVal input reads from the PDB file h2o.pdb:
molecule<Molecule>: ( pdb_file = "h2o.pdb" )
The following input explicitly gives the atom coordinates, using the ParsedKeyVal table notation:
molecule<Molecule>: (
unit=angstrom
{ atom_labels atoms geometry } = {
O1 O [ 0.000000000 0 0.369372944 ]
H1 H [ 0.783975899 0 -0.184686472 ]
H2 H [-0.783975899 0 -0.184686472 ]
}
)
)
The default units are Bohr with can be overridden with unit=angstrom#. The atom_labels# array can be omitted. The atoms# and geometry# arrays are required.
The Molecule class has a PointGroup member object, which also has a KeyVal constructor that is called when a Molecule is made. The following example constructs a molecule with 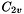 symmetry:
symmetry:
molecule<Molecule>: (
symmetry=c2v
unit=angstrom
{ atoms geometry } = {
O [0.000000000 0 0.369372944 ]
H [0.783975899 0 -0.184686472 ]
}
)
)
Only the symmetry unique atoms need to be specified. Nonunique atoms can be given too, however, numerical errors in the geometry specification can result in the generation of extra atoms so be careful.
|
|
Takes an (x, y, z) postion and finds an atom within the given tolerance distance. If no atom is found -1 is returned. |
|
|
Returns the index of the atom with the given label. If the label cannot be found -1 is returned. |
|
|
Converts an atom number to the number of its generating unique atom. The return value is in [0, nunique). |
|
|
Converts an atom number to the offset of this atom in the list of generated atoms. The unique atom itself is allows offset 0. |
|
|
Returns a double* containing the nuclear charges of the atoms. The caller is responsible for freeing the return value. |
|
|
This will try to carefully correct symmetry errors in molecules. If any atom is out of place by more then tol, abort will be called. |
|
|
Find this molecules true point group (limited to abelian groups). If the point group of this molecule is set to the highest point group, then the origin must first be set to the center of mass. |
|
|
Return 1 if this given axis is a symmetry element for the molecule. The direction vector must be a unit vector. |
|
|
Sets linear to 1 if the molecular is linear, 0 otherwise. Sets planar to 1 if the molecular is planar, 0 otherwise. |
|
|
Return 1 if the given plane is a symmetry element for the molecule. The perpendicular vector must be a unit vector. |
|
|
Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. This must be implemented by the derived class if the class has data. Reimplemented from SavableState. |
|
|
If the molecule contains only symmetry unique atoms, this function will generate the other, redundant atoms. The redundant atom will only be generated if there is no other atoms within a distance of tol. If the is another atom and it is not identical, then abort will be called. |