#include <wfn.h>
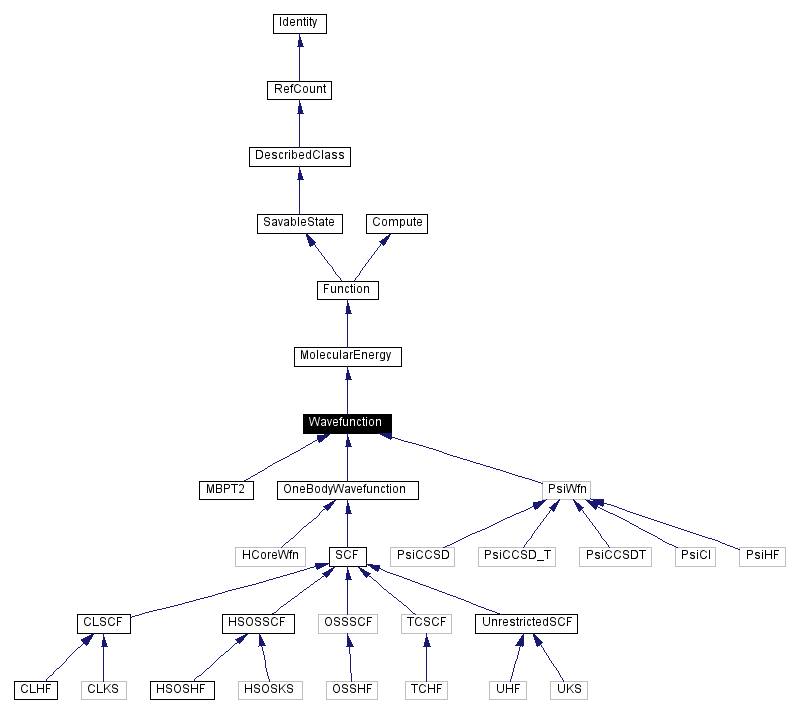
Inheritance diagram for Wavefunction
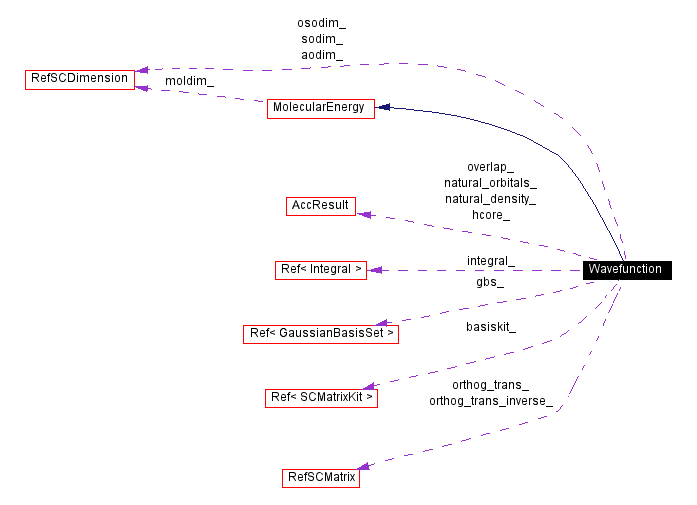
Public Types | |
| enum | OrthogMethod { Symmetric = 1, Canonical = 2, GramSchmidt = 3 } |
| An enum for the types of orthogonalization. | |
Public Methods | |
| Wavefunction (StateIn &) | |
| Wavefunction (const Ref< KeyVal > &) | |
| The KeyVal constructor. More... | |
| virtual | ~Wavefunction () |
| void | save_data_state (StateOut &) |
| Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. More... | |
| double | density (const SCVector3 &) |
| double | density_gradient (const SCVector3 &,double *) |
| double | natural_orbital (const SCVector3 &r, int iorb) |
| double | natural_orbital_density (const SCVector3 &r, int orb, double *orbval=0) |
| double | orbital (const SCVector3 &r, int iorb, const RefSCMatrix &orbs) |
| double | orbital_density (const SCVector3 &r, int iorb, const RefSCMatrix &orbs, double *orbval=0) |
| double | charge () |
| Returns the charge. | |
| virtual int | nelectron ()=0 |
| Returns the number of electrons. | |
| virtual RefSymmSCMatrix | density ()=0 |
| Returns the SO density. | |
| virtual RefSymmSCMatrix | ao_density () |
| Returns the AO density. | |
| virtual RefSCMatrix | natural_orbitals () |
| Returns the natural orbitals. | |
| virtual RefDiagSCMatrix | natural_density () |
| Returns the natural density (a diagonal matrix). | |
| virtual int | spin_polarized ()=0 |
| Return 1 if the alpha density is not equal to the beta density. | |
| virtual RefSymmSCMatrix | alpha_density () |
| Return alpha electron densities in the SO basis. | |
| virtual RefSymmSCMatrix | beta_density () |
| Return beta electron densities in the SO basis. | |
| virtual RefSymmSCMatrix | alpha_ao_density () |
| Return alpha electron densities in the AO basis. | |
| virtual RefSymmSCMatrix | beta_ao_density () |
| Return beta electron densities in the AO basis. | |
| virtual RefSCMatrix | nao (double *atom_charges=0) |
| returns the ao to nao transformation matrix. | |
| virtual RefSymmSCMatrix | overlap () |
| Returns the SO overlap matrix. | |
| virtual RefSymmSCMatrix | core_hamiltonian () |
| Returns the SO core Hamiltonian. | |
| RefSCDimension | ao_dimension () |
| Atomic orbital dimension. | |
| RefSCDimension | so_dimension () |
| Symmetry adapted orbital dimension. | |
| RefSCDimension | oso_dimension () |
| Orthogonalized symmetry adapted orbital dimension. | |
| Ref<SCMatrixKit> | basis_matrixkit () |
| Matrix kit for AO, SO, orthogonalized SO, and MO dimensioned matrices. | |
| Ref<GaussianBasisSet> | basis () const |
| Returns the basis set. | |
| Ref<Integral> | integral () |
| Returns the integral evaluator. | |
| void | symmetry_changed () |
| Call this if you have changed the molecular symmetry of the molecule contained by this MolecularEnergy. | |
| RefSCMatrix | so_to_orthog_so () |
| Returns a matrix which does the default transform from SO's to orthogonal SO's. More... | |
| RefSCMatrix | so_to_orthog_so_inverse () |
| Returns the inverse of the transformation returned by so_to_orthog_so. | |
| OrthogMethod | orthog_method () |
| Returns the orthogonalization method. | |
| void | obsolete () |
| Marks all results as being out of date. More... | |
| void | print (std::ostream &=ExEnv::out()) const |
| Print information about the object. | |
Protected Methods | |
| double | min_orthog_res () const |
| double | max_orthog_res () const |
| void | copy_orthog_info (const Ref< Wavefunction > &) |
Protected Attributes | |
| int | debug_ |
|
|
The KeyVal constructor.
|
|
|
Marks all results as being out of date. Any subsequent access to results will cause Compute::compute() to be called. Reimplemented from Compute. Reimplemented in MBPT2. |
|
|
Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. This must be implemented by the derived class if the class has data. Reimplemented from MolecularEnergy. Reimplemented in MBPT2, CLHF, CLSCF, HSOSHF, HSOSSCF, SCF, UnrestrictedSCF, and OneBodyWavefunction. |
|
|
Returns a matrix which does the default transform from SO's to orthogonal SO's.
This could be either the symmetric or canonical orthogonalization matrix. The row dimension is SO and the column dimension is ortho SO. An operator |
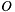 in the ortho SO basis is given by
in the ortho SO basis is given by 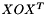 where
where 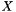 is the return value of this function.
is the return value of this function.