The rigid-body method minimizes the six rotational and
translational degrees of freedom for each specified group of
atoms; i.e., the groups of atoms are treated as rigid bodies.
The complete energy function 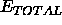 is used. The rotational
parameters are the three Eulerian angles
is used. The rotational
parameters are the three Eulerian angles 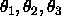 for a rotation around the geometric centers of the rigid group.
The three translational parameters are x,y,z in the Å frame.
Parts of the molecule that are not specified in any
GROUp statement will remain fixed. The constraints fix
statement (Section 8.1)
has no influence on rigid-body minimization (except
for the default group selection).
Upon completion of the last energy calculation,
symbols are declared that contain the
computed energy terms. The name of the symbols is given by
$<energy-term> (see Section
4.5). The overall energy (Eq. 4.1)
is stored in the symbol
$ENER; the rms gradient is stored in $GRAD. The value of
the second energy function (Eq. 4.26) is
returned in the symbol $PERT.
for a rotation around the geometric centers of the rigid group.
The three translational parameters are x,y,z in the Å frame.
Parts of the molecule that are not specified in any
GROUp statement will remain fixed. The constraints fix
statement (Section 8.1)
has no influence on rigid-body minimization (except
for the default group selection).
Upon completion of the last energy calculation,
symbols are declared that contain the
computed energy terms. The name of the symbols is given by
$<energy-term> (see Section
4.5). The overall energy (Eq. 4.1)
is stored in the symbol
$ENER; the rms gradient is stored in $GRAD. The value of
the second energy function (Eq. 4.26) is
returned in the symbol $PERT.