A bulk solvent mask calculation is available in X-PLOR.
The correction works by
defining the solvent region in the unit cell of the crystal, assigning an
average electron density to it, and Fourier-transforming this region. The
F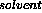 terms are stored in the FPART array and added to the calculated
structure factors (in FCALC) from the model being refined.
The FFT method is always used for this calculation.
terms are stored in the FPART array and added to the calculated
structure factors (in FCALC) from the model being refined.
The FFT method is always used for this calculation.
The solvent region is defined on the grid used for the model electron
density
generation for the FFT, which is initialized to the bulk solvent level
at each
point. By default, the bulk solvent density is 1.0
e Å
Å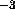 to simplify scaling (see below).
For each model atom, all grid points lying within
the sum of the half van der Waals radii of the model
atoms and an input solvent
radius are set to zero. After passing through all model atoms, the remaining
nonzero points define the solvent region. To avoid overcounting the solvent
points at the borders of the asymmetric units,
the entire unit cell is generated from this grid and the Fourier transform
computed in
to simplify scaling (see below).
For each model atom, all grid points lying within
the sum of the half van der Waals radii of the model
atoms and an input solvent
radius are set to zero. After passing through all model atoms, the remaining
nonzero points define the solvent region. To avoid overcounting the solvent
points at the borders of the asymmetric units,
the entire unit cell is generated from this grid and the Fourier transform
computed in  .
.