The distance geometry routines in X-PLOR begin by translating the bond lengths, bond angles, dihedral angles, improper angles, and van der Waals radii in the current molecular structure into a (sparse) matrix of distance bounds between the bonded atoms, atoms that are bonded to a common third atom, or atoms that are connected to each other through three bonds, as appropriate, using the equations in Crippen and Havel (1988). Experimental constraints can be added by the NOE assign statements (Section 18.1) and the restraints dihedral statements (Section 7.2). These lists of restraints are automatically read, translated into distance constraints, and entered into the bounds matrix.
The data translation is subject to the flag statement (Section 4.5); e.g., to include data from the distance restraint database, the following statement should be issued before invoking the mmdg statement :
FLAGsINCLude NOE END
The definition of upper and lower bounds proceeds in the following order: bond lengths, bond angles, dihedral angles, improper angles, distance and dihedral angle restraints, van der Waals repulsions, and rigid group distances. This order is actually not arbitrary; e.g., the calculation of 1--3 distance bounds from angles requires that distance bounds among the atoms used to define the parameter (in this case, bond lengths between atoms i and j and atoms j and k, as specified by the parameter statement) be defined previously.
For each bond, the accuracy parameter BACC is added to and subtracted from the equilibrium bond length as specified in the parameter statement (Section 3.2.1) or obtained from the reference coordinate set to give, respectively, the upper and lower bound on the particular 1--2 interatom distance.
Bond angle constraints are obtained from the parameter statement if REFErence=PARAmeter is specified or from the 1--3 distance obtained from the reference coordinate set if REFErence=COORdinates is specified. The accuracy parameter TACC is added to and subtracted from the equilibrium bond angle if REFErence=PARAmeter is specified, and BACC is added to and subtracted from the 1--3 distance if REFErence=COORdinates is specified.
Dihedral angle constraints
for single-minimum dihedral
angles defined by atoms i,j,k,l are
obtained from the parameter statement if REFErence=PARAmeter
is specified or from the distance between atoms i and l obtained from the
reference coordinate set if REFErence=COORdinates is specified.
The accuracy parameter PACC is
added to and subtracted from the equilibrium bond angle if
REFErence=PARAmeter is specified, and BACC is added to and subtracted
from the i,l distance if REFErence=COORdinates is specified.
For dihedral angles with multiple minima, the upper and lower
bounds correspond to the dihedral angle settings of 180 and 0
and 0 . In addition to reading all dihedral angles from the
parameter statement database, a generic list
comprising all possible dihedral angles in the
current molecular structure is generated. The upper and lower bounds
for these generic dihedral angles are set to 180
. In addition to reading all dihedral angles from the
parameter statement database, a generic list
comprising all possible dihedral angles in the
current molecular structure is generated. The upper and lower bounds
for these generic dihedral angles are set to 180 and 0
and 0 .
.
Improper angle constraints defined by atoms i,j,k,l are obtained from the parameter statement if REFErence=PARAmeter is specified or from the distance between atoms i,l obtained from the reference coordinate set if REFErence=COORdinates is specified. The accuracy parameter IACC is added to and subtracted from the equilibrium bond angle if REFErence=PARAmeter is specified, and BACC is added to and subtracted from the i,j distance if REFErence=COORdinates is specified.
For distance restraints
(see NOE statement,
Section 18.1), the current
distance database is read. Upper and lower bounds are
set to
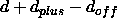 ,
, 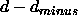 , where d,
, where d, 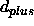 ,
and
,
and 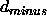 are specified through the NOE assign statements
and
are specified through the NOE assign statements
and 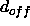 is specified through the NOE SQOFfset statement
(by default
is specified through the NOE SQOFfset statement
(by default 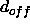 is zero).
is zero).
For dihedral angle restraints defined by atoms i,j,k,l (Section 7.2), the database is read as specified by the assign statements and interpreted as upper and lower bounds for the corresponding i,l distance.
To handle the repulsive van der Waals interactions , the nonbonded 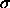 value
(see NONBonded statement in
Section 3.2.1) is converted into a van der Waals distance
(see Eq. 4.10) and then multiplied by the REPEl parameter as
part of the NBONd statement in the parameter statement
(see Section 3.2.1). All atom-atom repulsions are included
except for 1--3 and 1--2 atom interactions, regardless of
the NBXMOd parameter.
value
(see NONBonded statement in
Section 3.2.1) is converted into a van der Waals distance
(see Eq. 4.10) and then multiplied by the REPEl parameter as
part of the NBONd statement in the parameter statement
(see Section 3.2.1). All atom-atom repulsions are included
except for 1--3 and 1--2 atom interactions, regardless of
the NBXMOd parameter.
The group option calculates the distance between each pair of atoms in the group and enters it into the bounds matrix. It is important that the main coordinate set be well defined for the selected atoms. Multiple GROUp statements define multiple groups. The accuracy parameter specifies a value that will be added to and subtracted from the precise distance as obtained from the coordinates.