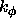 , which has units of
kcal mole
, which has units of
kcal mole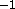 rad
rad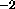 , and
the second real specifies
, and
the second real specifies 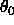 , the
equilibrium angle,
which has units of degrees (Eq. 4.5).
The optional UB specification activates the Urey-Bradley
term (Eq. 4.5), where the first real is the
Urey-Bradley energy constant
, the
equilibrium angle,
which has units of degrees (Eq. 4.5).
The optional UB specification activates the Urey-Bradley
term (Eq. 4.5), where the first real is the
Urey-Bradley energy constant 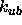 and the second real
is the Urey-Bradley equilibrium distance
and the second real
is the Urey-Bradley equilibrium distance 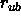 between the first
and the third atom that define the angle. If UB is not specified,
the Urey-Bradley equilibrium distance and energy constant default
to zero.
The program automatically
performs an interchange of the first with the third atom types
where this is required.
between the first
and the third atom that define the angle. If UB is not specified,
the Urey-Bradley equilibrium distance and energy constant default
to zero.
The program automatically
performs an interchange of the first with the third atom types
where this is required.
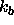 , which is the energy
constant in units of kcal mole
, which is the energy
constant in units of kcal mole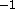 Å
Å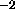 , and
the second real specifies
, and
the second real specifies
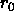 , which is the equilibrium bond length in Å
(Eq. 4.4).
The program automatically performs
an interchange of the two atom types
where this is required.
, which is the equilibrium bond length in Å
(Eq. 4.4).
The program automatically performs
an interchange of the two atom types
where this is required.
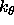 , the
integer is the periodicity n, and the
second real specifies
, the
integer is the periodicity n, and the
second real specifies  , the phase-shift angle, which
has units
of degrees. If the periodicity n is greater than 0, k has the
units of kcal mole
, the phase-shift angle, which
has units
of degrees. If the periodicity n is greater than 0, k has the
units of kcal mole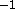 ; if the periodicity is 0, k has the units
of kcal mol
; if the periodicity is 0, k has the units
of kcal mol rad
rad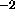 (Eq. 4.6).
The special character X is reserved for
the following combination: X <type> <type> X; it
acts as a wildcard. The
parameter retrieval
for a specified dihedral angle proceeds in the following way: first, a match
without wildcards is attempted, and second, a match against dihedral
parameters containing
wildcards is attempted. Some instances (such as sugars) require the mixing of
multiple dihedral angle terms with different periodicities.
In this case, dihedral statements with the multiple option
should be given in the parameter statement and
in the topology statement (Section 3.1.1).
Wildcards are
not allowed for multiple dihedral angles. The program automatically
performs the interchange
(a b c d)
(Eq. 4.6).
The special character X is reserved for
the following combination: X <type> <type> X; it
acts as a wildcard. The
parameter retrieval
for a specified dihedral angle proceeds in the following way: first, a match
without wildcards is attempted, and second, a match against dihedral
parameters containing
wildcards is attempted. Some instances (such as sugars) require the mixing of
multiple dihedral angle terms with different periodicities.
In this case, dihedral statements with the multiple option
should be given in the parameter statement and
in the topology statement (Section 3.1.1).
Wildcards are
not allowed for multiple dihedral angles. The program automatically
performs the interchange
(a b c d) 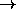 (d c b a) where this is required.
(d c b a) where this is required.
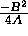 and the second is the distance
and the second is the distance
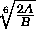 (Eq. 4.25).
(Eq. 4.25).
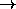 (a X X d)
(a X X d) 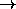 (X b c d)
(X b c d)
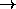 (X b c X)
(X b c X) 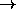 (X X c d).
The program automatically
performs the interchange
(a b c d)
(X X c d).
The program automatically
performs the interchange
(a b c d) 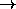 (d c b a) where this is required.
(d c b a) where this is required.
 5) nonbonded interactions.
Appropriate NONB statements have to be specified
for both atom types
before invoking this statement. The NBFIx statement allows
one to deviate from the standard combination rule for the
Lennard-Jones potential (Eq. 4.14).
Once an NBFIx statement has been issued for a single atom
type, say ``NBFIx HT HT", the combination rules for subsequent
NBONded statements will be affected, i.e., the ``NBFIx HT HT"
statement is equivalent to a ``NONBonded HT" statement.
5) nonbonded interactions.
Appropriate NONB statements have to be specified
for both atom types
before invoking this statement. The NBFIx statement allows
one to deviate from the standard combination rule for the
Lennard-Jones potential (Eq. 4.14).
Once an NBFIx statement has been issued for a single atom
type, say ``NBFIx HT HT", the combination rules for subsequent
NBONded statements will be affected, i.e., the ``NBFIx HT HT"
statement is equivalent to a ``NONBonded HT" statement.
NONB ( chemical C* ) 0.12 3.74 0.12 3.74 NONB ( chemical N* ) 0.24 2.85 0.24 2.85 NBFIx ( chemical C* ) ( chemical N* ) 10. 1000. 10. 1000.Note that the TOKEn keyword is not allowed for the reals. Once an NBFIx statement has been issued for a single atom type, say ``NBFIx (type HT) (type HT)", the combination rules for subsequent NBONded statements will be affected, i.e., the ``NBFIx (type HT) (type HT)" statement is equivalent to a ``NONBonded (type HT)" statement.
 ,
,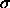 (Eq. 4.8)
for all nonbonded interactions except the
special 1--4 interactions; the second pair
is
(Eq. 4.8)
for all nonbonded interactions except the
special 1--4 interactions; the second pair
is  ,
,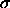 for the 1--4 nonbonded
interactions (NBXMod=
for the 1--4 nonbonded
interactions (NBXMod= 5).
5).
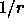 -dependent
dielectric (Eq. 4.16). RDIE may
only be used in combination with VSWItch, SWITch, and REPEl=0.
CDIE may be used in combination with VSWItch, SHIFt, and REPEl=0
or in combination with TRUNcation and REPEl=0 (default: CDIE).
-dependent
dielectric (Eq. 4.16). RDIE may
only be used in combination with VSWItch, SWITch, and REPEl=0.
CDIE may be used in combination with VSWItch, SHIFt, and REPEl=0
or in combination with TRUNcation and REPEl=0 (default: CDIE).
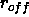 at which the
switching function or shifting function
forces the nonbonded energy to zero (Eqs. 4.8,
4.16) (default: 7.5 Å).
at which the
switching function or shifting function
forces the nonbonded energy to zero (Eqs. 4.8,
4.16) (default: 7.5 Å).
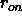 at
which the switching
function becomes effective (Eq. 4.8) (default: 6.5 Å).
at
which the switching
function becomes effective (Eq. 4.8) (default: 6.5 Å).
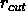 for
the nonbonded list generation (default: 8.5 Å).
for
the nonbonded list generation (default: 8.5 Å).
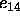 for the special
1--4 electrostatic interactions (Eq. 4.17)
(default: 1.0).
for the special
1--4 electrostatic interactions (Eq. 4.17)
(default: 1.0).
 (Eq. 4.16) (default: 1.0).
(Eq. 4.16) (default: 1.0).
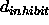 (Eq. 4.7) between two atoms
below which the van der Waals potential
(Eq. 4.8) is truncated (default: 0.25 Å).
(Eq. 4.7) between two atoms
below which the van der Waals potential
(Eq. 4.8) is truncated (default: 0.25 Å).
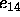 (Eqs. 4.17 and 4.18).
(Eqs. 4.17 and 4.18).
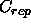 for
the repel function (Eq. 4.8)
(default: 100.0).
for
the repel function (Eq. 4.8)
(default: 100.0).
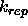 : if > 0, this option
turns on the repel function (Eq. 4.8) and turns off the
electrostatic energy.
: if > 0, this option
turns on the repel function (Eq. 4.8) and turns off the
electrostatic energy. 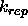 specifies the factor by which
to multiply the van der Waals radius
specifies the factor by which
to multiply the van der Waals radius 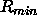 (default: 0).
(default: 0).
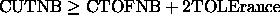 . In this
way the nonbonded energy is independent of the update frequency.
For the REPEl option, CUTNB and TOLErance should be chosen
such that
. In this
way the nonbonded energy is independent of the update frequency.
For the REPEl option, CUTNB and TOLErance should be chosen
such that 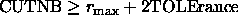 ,
where
,
where 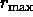 is the maximum van der Waals radius.
TOLErance has no influence on the TRUNcation option.
(default: 0.5 Å ).
is the maximum van der Waals radius.
TOLErance has no influence on the TRUNcation option.
(default: 0.5 Å ).
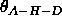 (Eq. 4.25).
(Eq. 4.25). 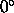 corresponds to a linear hydrogen bond
(default:
corresponds to a linear hydrogen bond
(default: 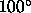 ).
).
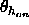 , a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default:
, a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default: 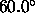 ).
).
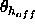 ,
a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default:
,
a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default: 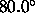 ).
).
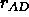 , the heavy atom
donor to heavy atom acceptor cutoff (Eq. 4.25)
(default: 7.5).
, the heavy atom
donor to heavy atom acceptor cutoff (Eq. 4.25)
(default: 7.5).
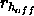 ,
a switching function parameter for the hydrogen bond
distance (Eq. 4.25) (default: 6.5).
,
a switching function parameter for the hydrogen bond
distance (Eq. 4.25) (default: 6.5).
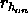 , a
switching function parameter for the hydrogen bond distance
(Eq. 4.25) (default: 5.5).
, a
switching function parameter for the hydrogen bond distance
(Eq. 4.25) (default: 5.5).
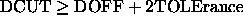 . In this
way the hydrogen-bonded
energy is independent of the update frequency (default: 0.5).
. In this
way the hydrogen-bonded
energy is independent of the update frequency (default: 0.5).