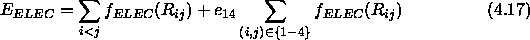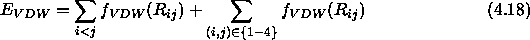The intramolecular interaction energy is the summation of the individual nonbonded interaction energies for pairs of atoms within the current molecular structure, e.g., a single molecule or a crystallographic asymmetric unit.
The summation extends over all pairs of atoms (i<j) that satisfy the cutoff criteria specified by CUTNb and ATOM or GROUP ( nbonds statement, see Section 3.2.1) and that are selected by the constraints interaction statement. The program's interpretation of the cutoff points depends on the type of cutoff used. For the group cutoff option, the program first executes a group-by-group centroid search to determine which centroids are within the particular cutoff value CUTNb of each other. Then the program calculates the energy using every atom in both groups. For the atom cutoff, the nonbonded interactions are included on an atom-by-atom basis. However, the pairwise search is computationally expensive. A great reduction in computational time is achieved by first searching for nearest neighbors among the nonbonded groups and then searching for atom pairs between selected group pairs.
The computational time is further reduced
by introducing an approximation, storing the atomic pair
indices (i<j) that satisfy 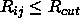 in a
list that is updated
only when any atom has moved more than the amount specified by
TOLErance. For both switched and shifted nonbonded
options (see Section 3.2.1),
the distance
in a
list that is updated
only when any atom has moved more than the amount specified by
TOLErance. For both switched and shifted nonbonded
options (see Section 3.2.1),
the distance 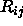 at which the energy becomes
zero is given by
at which the energy becomes
zero is given by 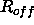 . Thus, the nonbonded energy
calculations become independent of the update frequencies if
. Thus, the nonbonded energy
calculations become independent of the update frequencies if
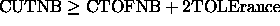 .
.
There are a number of cases for nonbonded interactions that
must not be computed, e.g., interactions between covalently bonded
atoms. Covalently bonded exclusions are automatically generated
by X-PLOR using information about atom connectivity.
In addition, certain exclusions can be added manually by
the EXCLude statement, which is an atom statement
(see Section 3.1.1).
The NBXMod statement
(see Section 3.2.1)
has several options for automatically excluding
1--2, 1--2 and 1--3, and 1--2, 1--3,
and 1--4 interactions in the molecule. In the case of
NBXMod= 5, the 1--4 interactions are treated in a special way.
The electrostatic 1--4 interactions are scaled by
5, the 1--4 interactions are treated in a special way.
The electrostatic 1--4 interactions are scaled by 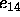 , and the
van der Waals interactions use a special 1--4 set of parameters for
, and the
van der Waals interactions use a special 1--4 set of parameters for
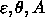 and B. In the case of
NBXMod
and B. In the case of
NBXMod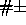 5, 1--4 interactions are treated as normal nonbonded
interactions, and the second terms of the right-hand side of
Eqs. 4.17 and 4.18 become zero.
5, 1--4 interactions are treated as normal nonbonded
interactions, and the second terms of the right-hand side of
Eqs. 4.17 and 4.18 become zero.